
Introduction
Single cell whole genome bisulfite sequencing (scwgbs) is based on the whole genome DNA at the single cell level, which is treated with bisulfite and combined with high-throughput sequencing technology to study the whole genome methylation level of a single cell. The result will accurately analyze the methylation status of each cytosine in a single cell. The methylation research of single cell and trace rare samples is mainly applied to the research fields of tumorigenesis mechanism, cancer research, preimplantation diagnosis, early embryonic development, germ cell recombination, stem cells and cell heterogeneity.
Technical advantages
1.Ultra low starting quantity
Single cell or ultra-low starting amount of DNA for Library Construction
2.测序覆盖度高
Maximize access to complete genome-wide methylation information and accurately map methylation
3. single base resolution
It can accurately analyze the methylation status of each C base
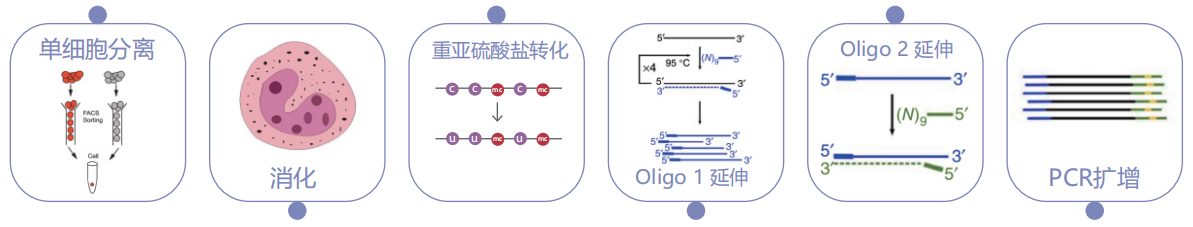
Experimental strategy
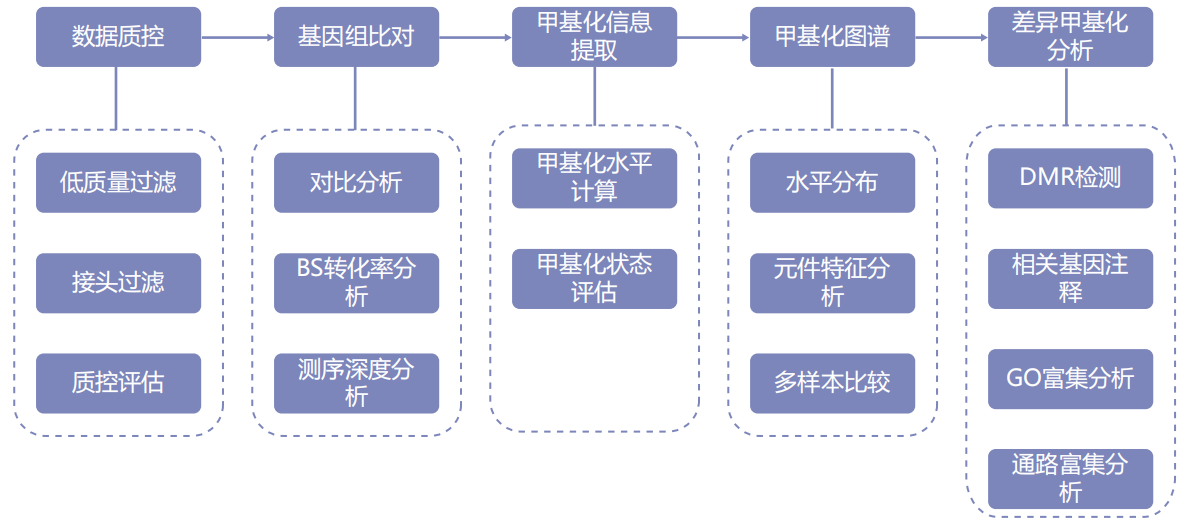
Information analysis
Technical parameter
| Sample requirements | Project cycle |
| Cell isolation: the partner shall complete the single-cell isolation sample requirements: single cell, a small number of cells and other rare samples or genomic DNA amplification technology of more than 5 pg: random primer amplification technology sequencing depth: ≥ 30x | The standard operation cycle below 15 samples is about 60 working days. Because different species require different amounts of data, and the number of databases required varies greatly, the project cycle needs to be determined with the technical support personnel in case of a large number of samples. |
Case analysis
Case: single cell DNA methylome Atlas of human preimplantation embryo development
Zhu, P., et al. (2018). Single-cell DNA methylome sequencing of human preimplantation embryos. Nat Genet 50(1).
Background:
In mammalian genomes, cytosines (mainly cytosines in CpG dyads) are methylated under the catalysis of DNA methylases. Studies have shown that DNA methylation is essential for many biological processes, such as gene expression repression, transposon transcriptional activity regulation, X chromosome inactivation, and genome imprinting maintenance. Previous studies have shown that there is only large-scale DNA demethylation during early embryonic development before implantation. According to the data of this study, after sperm and egg cells are combined for fertilization, while large-scale DNA demethylation occurs in human early embryos, a large number of highly specific DNA is also de novo methylated, which indicates that during the first round of DNA methylome reprogramming in human early embryos, the overall 'net result' of DNA demethylation is actually the result of dynamic balance generated by the antagonism between the two molecular processes of highly ordered large-scale DNA demethylation and local DNA plus methylation.
Methods:
Using the high-throughput sequencing method of single-cell DNA methylome, we have carried out a more in-depth analysis of human preimplantation embryo development process at single-cell resolution for the first time
Conclusion:
In this article, in order to further study the dynamic characteristics of DNA methylation reprogramming process at the single-cell level, we used single-cell genome-wide DNA methylome high-throughput sequencing technology to systematically study the single-cell and single base discrimination rate at each key stage of human preimplantation embryo development. The main findings are as follows: (1) for the first time, we found that there are a large number of specific de novo DNA methylation during human preimplantation embryo development. (2) It was found for the first time that the remaining methylation level on the parental genome was reversed from the two cell embryonic stage, and the remaining methylation level on the maternal genome was significantly higher than that on the paternal genome in the same single cell. (3) It is the first time to find that the asymmetric allocation of DNA methylation during early embryonic cleavage can be used to trace the genetic lineage of each cell in the same embryo.
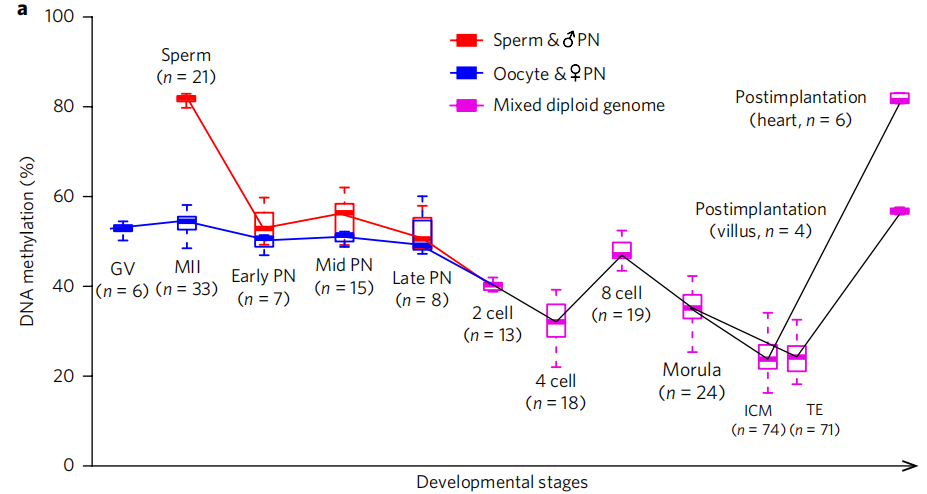
植入前胚胎不同发育阶段DNA甲基化的动态变化

