DNA&蛋白亲和纯化测序(DAP-seq):(DNA affinity purification sequencing) 是一种高通量 TF 结合位点发现方法,用 体外表达的 TFS 来询问基因组 DNA. DAP-seq 数据集还可以了解到许多 TFS 的生物学和绑 定站点体系结构,(一种高通量的检测方法,使用体外表达的 TF 来询问裸露的 gdna 片段, 以建立结合位置 (峰) 和序列基序。)DAP-seq 是一种体外 TF-dna 结合试验,它允许低成本 快速生成大量 tfs 的全基因组结合位点图,同时捕获影响体内结合的 gdna 特性。
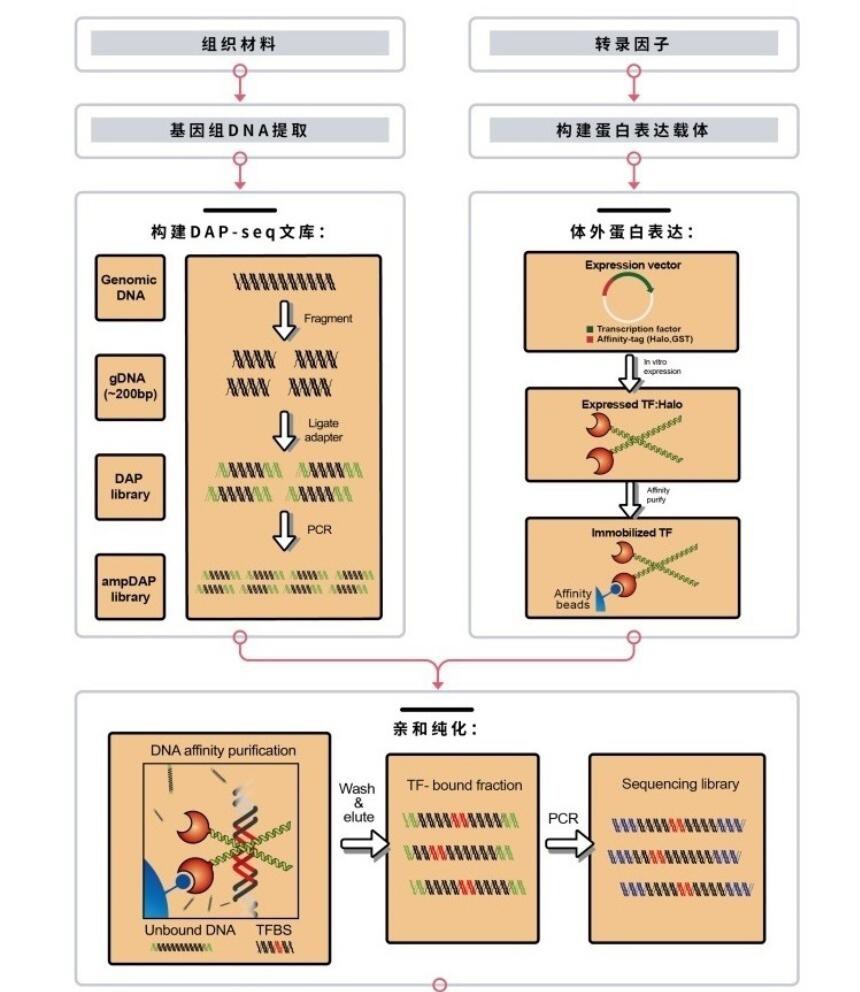
图 1.1: DAP-Seq 技术流程原理
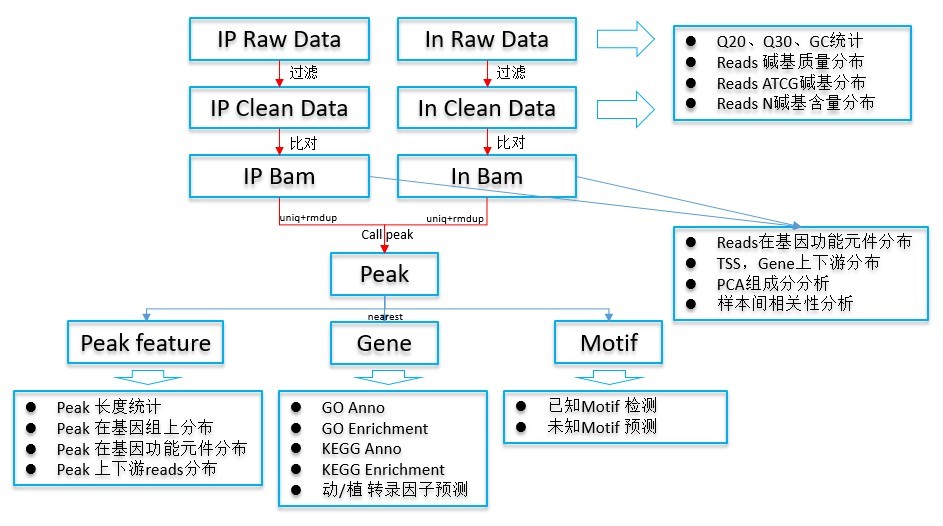
图 2.1: DAP-seq 信息分析流程
信息分析主要分为 5 部分: 第一部分,数据预处理。去接头序列、污染序列、低质量碱基,获得 clean data 序列,并
进行相关数据统计;
第二部分,clean data 定位到参考基因组上,得到 bam 文件,并去除重复序列,保留唯 一比对的序列;
第三部分,将 bam 文件进行 Peak 检测,得到富集区域的信息,并进行 Peak 在基因功 能元件的分布,最近基因寻找及 motif 预测。
第四部分,统计 Peak 分布情况,对 Peak 最近基因进行 GO、KEGG 功能注释与富集及 转录因子预测等。
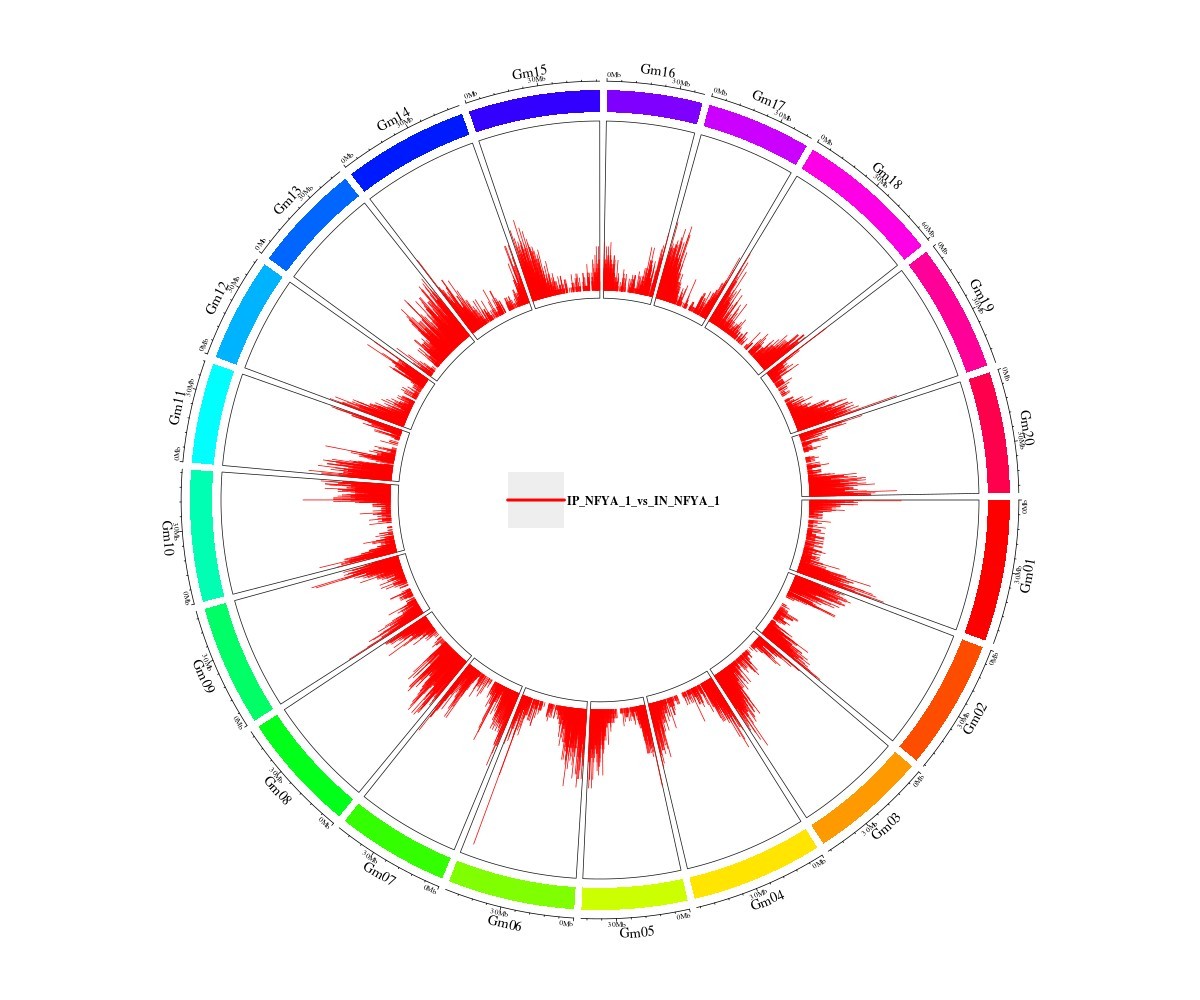
富集区间分布特征
根据基因组大小,设定动态长度的窗口,统计每个窗口内 Peak 的数量,并绘图展示 Peak
在基因组上的分布
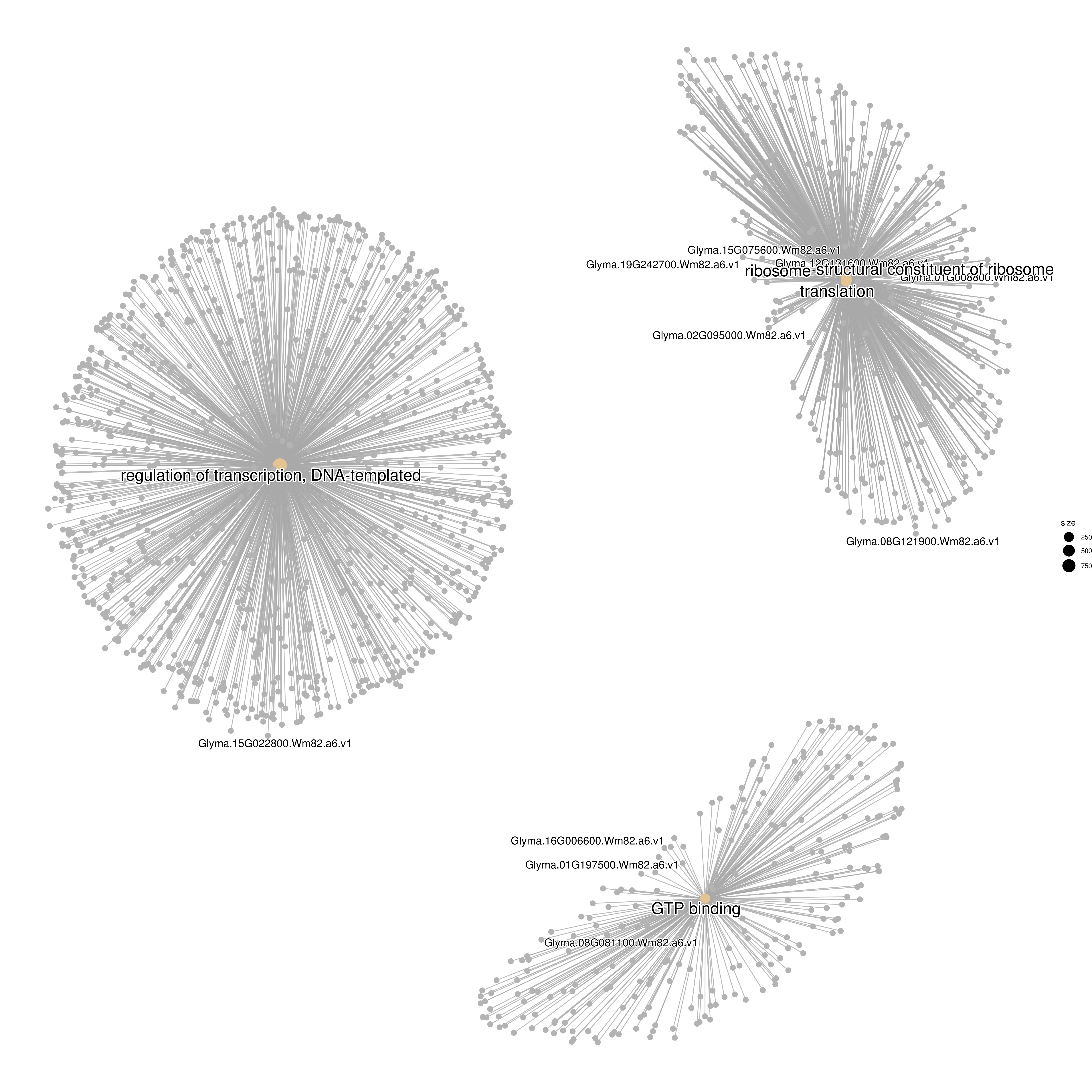
Peak 关联基因的 GO 富集网络图 将所有 GO 富集结果,按照 pvalue 从小到大排序,取前 10 个 GO 富集功能绘制网络图
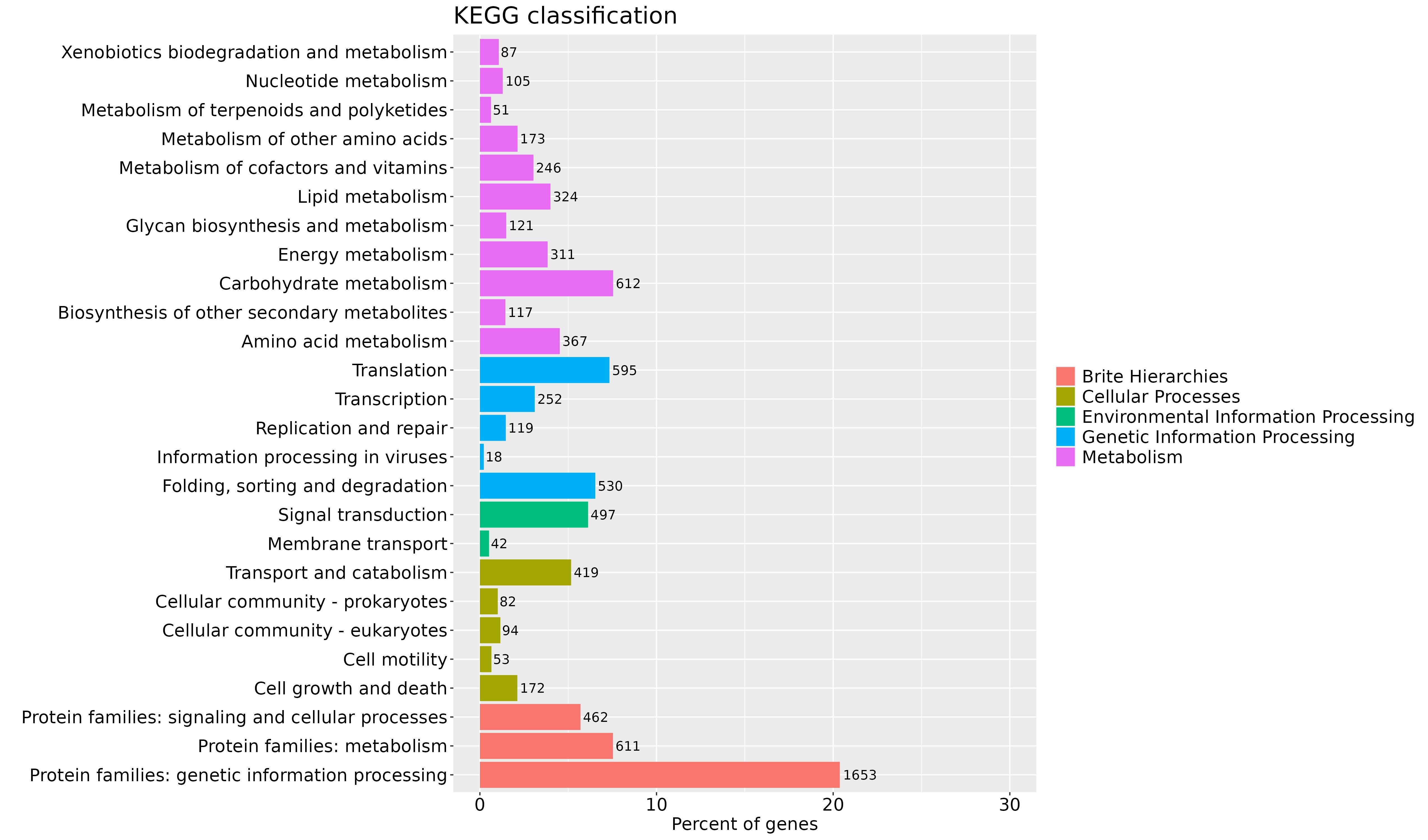
Peak 关联基因的 KEGG 聚类
将 KEGG 注释对应的 gene 数目,进行统计然后功能分类绘图
富集区间 Motif 分析 Motif 是具有某种生物特性,在一组相关序列中重复出现的序列模式,是研究基因表达 调控机制的重要信息。DAP-seq 所研究的蛋白 (转录因子),其结合位点可能存在特定的序列 特征。为尝试得到这种修饰结合的保守序列信息 (motif 序列),进一步理解蛋白修饰对基因 表达的调控,在得到富集区间 (Peak) 之后将通过 Homer 软件(version 3) 预测 motif,将所有的 Peak 序列用于预测 motif。以下是部分预测结果

参考文献
[1] Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data.a
[2] Bolger A M , Lohse M , Usadel B . Trimmomatic: a flexible trimmer for Illumina sequence data[J]. Bioinformatics, 2014, 30(15):2114-2120.
[3] Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25:1754-60.
[4] Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A., & Manke, T. (2014). Deeptools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Research, 42(Web Server issue), 187-91.
[5] ZHANG, Yong, et al. Model-based analysis of ChIP-Seq (MACS).?Genome biology, 2008, 9.9: R137.
[6] KRZYWINSKI, Martin, et al. Circos: an information aesthetic for comparative genomics.
Genome research, 2009, 19.9: 1639-1645.
[7] Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, & Cherry JM, et al. (2000). Gene ontology: tool for the unification of biology. the gene ontology consortium.?Nature Genetics,?25(1), 25-9.
[8] Kanehisa, M., & Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research, 28(1), 27-30.
[9] Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcrip- tion factors prime cis-regulatory elements required for macrophage and B cell identities[J]. Molecular cell, 2010, 38(4): 576-589.
[10] Stark, R., & Brown, G. (2011). DiffBind: differential binding analysis of ChIP-Seq Peak data. R package version, 100, 4-3.
[11] Zheng, Y., Jiao, C., Sun, H., Rosli, H. G., Pombo, M. A., & Zhang, P., et al. (2016). Itak: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Molecular Plant, 9(12), 1667-1670.